Product Sheet CA1317
Description
BACKGROUND
Cytochrome c oxidase (EC 1.9.3.1) (COX) belongs to the superfamily of terminal oxidases present in all aerobic organisms. In eukaryotic cells, cytochrome c oxidase is embedded as a dimer in the mitochondrial inner membrane. Electron transfer by the enzyme from cytochrome c to molecular oxygen is coupled to proton pumping across the membrane, contributing to the mitochondrial membrane potential, resulting in a proton and charge gradient that is then employed by the F0F1-ATPase to synthesize ATP. The redox centers involved in electron transfer are two copper centers (CuA and CuB) and two heme A moieties (a and a3). The eukaryotic enzyme contains three mitochondrial DNA (mtDNA)-encoded subunits: MTCO1, MTCO2, and MTCO3. These subunits are homologous to polypeptides found in bacterial cytochrome c oxidases. They form both the catalytic and structural core of the enzyme. The hydrophobic interior of enzyme subunit 1 (MTCO1) coordinates the heme a group and a binuclear center composed of heme a3 and CuB. The intermembrane space domain of enzyme subunit 2 (MTCO2) coordinates the binuclear CuA center. In addition to the three mtDNA-encoded subunits, mammalian cytochrome c oxidase contains 10 nuclear DNA-encoded subunits: COX4, COX5A, COX5B, COX6A, COX6B, COX6C, COX7A, COX7B, COX7C, and COX8. In humans, COX6A and COX7A exist as skeletal/cardiac muscle (COX6A2 and COX7A1) and ubiquitously expressed (COX6A1 and COX7A2) isoforms. Although not confirmed at the protein level, human isoforms of COX4, COX6B, and COX8 have also been identified.1
Cytochrome c oxidase (COX) deficiencies are one of the most common defects of the respiratory chain found in mitochondrial diseases. Cytochrome c oxidase (COX) biosynthesis requires numerous assembly factors that do not form part of the final complex but participate in prosthetic group synthesis and metal delivery in addition to membrane insertion and maturation of cytochrome c oxidase (COX) subunits. Human diseases associated with cytochrome c oxidase (COX) deficiency including encephalomyopathies, Leigh syndrome, hypertrophic cardiomyopathies, and fatal lactic acidosis are caused by mutations in cytochrome c oxidase (COX) subunits or assembly factors. Owing to the importance of the enzyme, pathogenetic mutations affecting cytochrome c oxidase (COX) frequently result in severe, often fatal metabolic disorders.2 In the last decade, numerous animal models have been created to understand the pathophysiology of cytochrome c oxidase (COX) deficiencies and the function of assembly factors. These animal models, ranging from invertebrates to mammals, in most cases mimic the pathological features of the human diseases.3
References:
1. Capaldi, R.A.: Annu. Rev. Biochem. 59: 569-596, 1990
2. Pecina, P. et al: Physiol. Res. 53(Suppl. 1):S213-S223, 2004
3. Diaz, F.: Biochim Biophys Acta. 1802:100-10, 2010
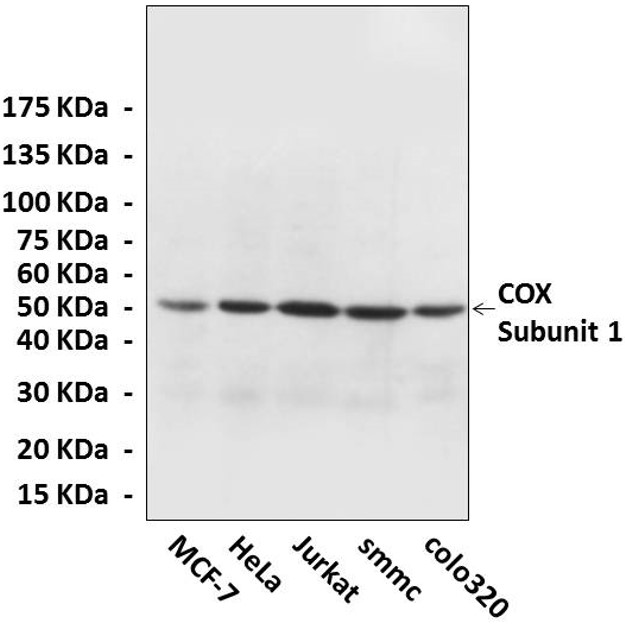
(Click to Enlarge) Western Blot detection of cytochrome c oxidase proteins in various cell lysate using cytochrome c oxidase Antibody.
Details
| Cat.No.: | CA1314 |
| Antigen: | Short peptide from cytochrome c oxidase subunit 1 sequence. |
| Isotype: | Affinity-Purified Rabbit Polyclonal IgG |
Species & predicted species cross- reactivity ( ): | Human |
Applications & Suggested starting dilutions:* | WB 1:1000 IP n/d IHC (Paraffin) 1:50-200 ICC n/d FACS n/d |
Predicted Molecular Weight of protein: | 50 kDa |
| Specificity/Sensitivity: | Detects endogenous levels of cytochrome c oxidase subunit 1 proteins without cross-reactivity with other related proteins. |
| Storage: | Store at -20°C for at least one year. Store at 4°C for frequent use. Avoid repeated freeze-thaw cycles. |
*Optimal working dilutions must be determined by end user.